
Recent Developments in Roundup Cancer Trials As of early 2024, we've witnessed an unprecedented surge in the legal...
Experienced Lawyers and Complex Litigation Attorneys
Class action lawsuits represent one of the last and most powerful means of reigning in corporate wrongdoing. Our blog presents information on current and past class actions, as well as noteworthy developments in major class action litigation throughout the United States.
Recent Developments in Roundup Cancer Trials As of early 2024, we've witnessed an unprecedented surge in the legal...

Macy's Bedsheet Count Financial Settlement News An Ohio federal judge, Douglas R. Cole, denied final approval of a...
Significant Developments in 3M Earplug Multidistrict Litigation A federal judge in Florida, overseeing the 3M earplug multidistrict litigation...
Missouri Jury Sides with Home Sellers in a Landmark Class Action Case In a landmark verdict, a jury...
Cancer Survivor Concludes Case Against Monsanto Over Roundup's Alleged Link to Lymphoma Mark McCostlin, a cancer survivor, rested...
Walgreens Settles Theranos Blood Test Class Action for $44 Million Walgreens has consented to a $44 million settlement...
Class Action Law Update - August 21, 2023 Recent Trends in Class Action Litigation and Notable Settlements Class...
Class Action Law Update - August 7, 2023 $10.5 Million Settlement for Macy's Bedsheet Buyers Three buyers who...
Class Action Law Update - July 12, 2023 Philips Calls for Dismissal of Claims in Multidistrict Litigation Philips...
Class Action Law Update - May 26, 2023 Arbitration Agreements Gain Traction in Automotive Industry Automakers are increasingly...
Class Action Law Update - April 24, 2023 Several noteworthy class action lawsuits have recently been filed across...
Class Action Update - February 1, 2023 Apple accused of tracking user data of users who have opted out...
Class Action Case Update – December 16, 2022 Facebook algorithms accused of age and gender discrimination while displaying...
Class Action Case Update – November 25, 2022 Veterans accuse Citibank of non-compliance with servicemen-related lending requirements A...
Class Action Case Update - October 24, 2022 Amplify settlement in 2021 Huntington oil spill within sight -...
Class Action Case Update – October 13, 2022 Illegal tracking lawsuit: Google to pay $85 million to the...
Class Action Case Update - September 28, 2022 Crypto companies facing regulatory wrath combined with investor wrath The...
Class Action Case Update - September 14, 2022 Credit Karma to pay USD 3 million for luring consumers...
Top Universities’ motion to dismiss financial aid case is denied A total of 17 private schools (including Yale,...
Meta accused of knowingly employing exploitative and damaging business tactics so as to lure young users A total...
Court rules against Whole Foods in a preliminary motion seeking dismissal of a suit alleging the latter of...
Subway Loses Bid to Throw Out Lawsuit Based on Ingredients in Tuna Sandwiches A federal court judge has...

Audet & Partners, LLP is investigating an increasing number of complaints as part of a TracFone lawsuit that...

Audet & Partners, LLP is investigating claims as part of a John Deere lawsuit that Deere has illegally...

Audet & Partners, LLP has initiated a FedEx ADA class action in the United States District Court for...

Audet and Partners, LLP is investigating claims as part of a Peloton lawsuit that purchasers of the high-end...

Audet & Partners, LLP is pursuing claims as part of a comedian class action that prominent streaming services,...

Audet and Partners, LLP is investigating claims by an increasing number of consumers as part of a Canon...

July 2022 Update: The Huntington Beach Oil Spill has been settled. Please click here to view more information...

Audet & Partners, LLP is investigating claims as part of a Philips CPAP lawsuit on behalf of individuals...

Audet & Partners, LLP is investigating an increasing number of consumer complaints that have surfaced alleging false health...

Audet and Partners, LLP is investigating claims as part of an infant formula lawsuit on behalf of purchasers...

Audet and Partners, LLP is investigating an increasing number of consumer complaints as part of a child booster...

Are you a Fortnite player and use an iPhone? As you may be aware, Apple Inc. has advised...

Audet & Partners, LLP is investigating claims by purchasers of Generac generators that the Generac's Guardian Series generators were...

Audet & Partners, LLP is investigating claims that may form the basis of a class action lawsuit against...

A lawsuit brought on behalf of more than 30,000 Arizona prison inmates has been allowed to proceed as...

A judge in Nevada has granted class action certification in the lawsuit against Payday King Carey V. Brown,...

A lawsuit in California claims Intuitive Surgical, Inc., the maker of the da Vinci Surgical System, misled shareholders...

Effective January 10, 2014, mortgage servicers are required to provide substantial new notice to consumers before charging any...
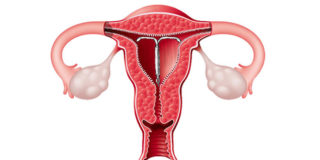
Given the increased use of IUDs as a form of long-term birth control, specifically the Mirena IUD, the...

Over objections raised by Bayer Healthcare Pharmaceuticals*, the United States Judicial Panel ("USJP") on Multidistrict Litigation has consolidated...

Bayer Healthcare Pharmaceuticals has taken a position opposing consolidation of legal actions brought against the medical device manufacturer...
A recent national study suggests that nearly 75% of cell phone users that send and receive text messages have received unsolicited or "spam" text messages advertising or promoting products and/or services. Another study estimates that the number of unique commercial text messaging ad campaigns quadrupled in the first half of 2012, and that receipt of such messages increased 300% between 2011 and 2012. Consumers are protected from unsolicited commercial text messages by the Federal Act known as the...

“A bank has an equitable right to set off a matured debt, owed the bank by a depositor,...
As attorneys, and as members of our communities, we feel that it is important that all people have access to legal resources. One issue has been that many folks simply do not know what to do, or who to contact, should they need legal help. We are not an attorney referral service, however, we do often refer cases out to other qualified lawyers and firms, if the case is not something that our firm handles. As part of our commitment to educating the...